Problem
To allow for the presence of its varying forms, a protein motif is represented by a shorthand as follows: [XY] means "either X or Y" and {X} means "any amino acid except X." For example, the N-glycosylation motif is written as N{P}[ST]{P}.
You can see the complete description and features of a particular protein by its access ID "uniprot_id" in the UniProt database, by inserting the ID number into
http://www.uniprot.org/uniprot/uniprot_id
Alternatively, you can obtain a protein sequence in FASTA format by following
http://www.uniprot.org/uniprot/uniprot_id.fasta
For example, the data for protein B5ZC00 can be found at http://www.uniprot.org/uniprot/B5ZC00.
Given: At most 15 UniProt Protein Database access IDs.
Return: For each protein possessing the N-glycosylation motif, output its given access ID followed by a list of locations in the protein string where the motif can be found.
Sample Dataset
A2Z669
B5ZC00
P07204_TRBM_HUMAN
P20840_SAG1_YEAST
Sample Output
B5ZC00
85 118 142 306 395
P07204_TRBM_HUMAN
47 115 116 382 409
P20840_SAG1_YEAST
79 109 135 248 306 348 364 402 485 501 614
Problem explanation
단백질 Access ID를 통해 uniprot에 접속하여 단백질 정보를 얻어와 특정 motif를 찾는 문제입니다. Dataset을 보아 크롤링이 필요할 것으로 추측되어 이용 했습니다. 다만 pre태크로 싸여있어 텍스트 파일로 저장한 후 다시 불러오는 방식(저의 크롤링 실력이 부족하여 방법을 못찾았을 수도 있습니다.)으로 진행했습니다. 또한 정규식을 이용하여 각 문자열에 있는 N-glycosylation motif를 찾았습니다.
Source
from bs4 import BeautifulSoup
import requests
import os
import re
def get_html(url):
_html = ""
resp = requests.get(url)
if resp.status_code == 200:
_html = resp.text
return _html
rosadirect = "D:/gs/rosalind/"
id, seq, start, total =[], [], [], []
f = open(rosadirect + "rosalind_mprt.txt","r")
lines = f.readlines()
for line in lines:
id.append(line.replace("\n", ""))
f.close()
p = re.compile(r"N[^P](S|T)[^P]")
for ide in id :
URL = "https://www.uniprot.org/uniprot/%s.fasta" % ide
html = get_html(URL)
soup = BeautifulSoup(html, 'html.parser')
f = open(rosadirect + "temp.txt", "w")
f.write(soup.getText())
f.close()
f = open(rosadirect + "temp.txt", "r")
lines = f.readlines()
seq= ""
totemp=[]
for j in range(1,len(lines)):
seq = seq + lines[j].replace("\n","")
for k in range(len(seq)+1):
a = seq[k:]
m = p.match(a)
if m is not None:
totemp.append(k+1)
if totemp == [[]]:
total.append('None')
else:
total.append(totemp)
f.close()
total = list(map(str,total))
for z in range(len(id)):
if total[z] == '[]':
continue
else:
print(id[z])
print(total[z].replace("[","").replace("]","").replace(",",""))
os.remove(rosadirect+"temp.txt")
Motif Implies Functionclick to collapse
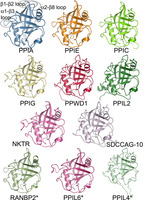
Figure 1. The human cyclophilin family, as represented by the structures of the isomerase domains of some of its members.
As mentioned in “Translating RNA into Protein”, proteins perform every practical function in the cell. A structural and functional unit of the protein is a protein domain: in terms of the protein's primary structure, the domain is an interval of amino acids that can evolve and function independently.
Each domain usually corresponds to a single function of the protein (e.g., binding the protein to DNA, creating or breaking specific chemical bonds, etc.). Some proteins, such as myoglobin and the Cytochrome complex, have only one domain, but many proteins are multifunctional and therefore possess several domains. It is even possible to artificially fuse different domains into a protein molecule with definite properties, creating a chimeric protein.
Just like species, proteins can evolve, forming homologous groups called protein families. Proteins from one family usually have the same set of domains, performing similar functions; see Figure 1.
A component of a domain essential for its function is called a motif, a term that in general has the same meaning as it does in nucleic acids, although many other terms are also used (blocks, signatures, fingerprints, etc.) Usually protein motifs are evolutionarily conservative, meaning that they appear without much change in different species.
Proteins are identified in different labs around the world and gathered into freely accessible databases. A central repository for protein data is UniProt, which provides detailed protein annotation, including function description, domain structure, and post-translational modifications. UniProt also supports protein similarity search, taxonomy analysis, and literature citations.
